Since he was only four weeks old, Daniel Nevins-Selvadurai has been called every synonym for a medical mystery.
At that young age, his mother brought him to see a doctor, suspecting that her baby simply had the flu. That doctor’s visit quickly turned into an ambulance ride to Toronto’s Hospital for Sick Children (SickKids), where an MRI confirmed he had a stroke.
Daniel made a full recovery from the stroke, but what triggered it, along with the growing list of other painful symptoms he experienced, remained a mystery.
Daniel had a weak immune system. He had stomach problems and blood in his stool, but he didn’t fit the genetic profile for inflammatory bowel disease. His father noticed that when he caught infections, they often led to rashes and skin lesions, and even sepsis.
As time passed, Daniel was referred to multiple clinical teams across many specialties, including hematology, oncology, rheumatology, immunology, gastroenterology, and dermatology. But doctors simply couldn’t explain Daniel’s condition. They felt certain that there was a genetic cause, but could not point to one.
This all started in 2006.
Dr. Aleixo Muise, a gastroenterologist at SickKids, and a leader in the study that would ultimately give Daniel a diagnosis 10 years later, recalls his frustration knowing that without a cause, the best he and his team could do was treat symptoms as they came up.
When Muise obtained a grant to sequence the genomes of several patients with inflammatory bowel disease and similar conditions, he immediately thought of Daniel and enrolled him in the study in hopes of finding answers.
The study, published in Nature Communications, revealed a previously undiscovered genetic mutation in a gene that was previously assumed to be essential to survival.
Daniel had an ARPC1B mutation, which normally produces a core protein of the Arp2/3 complex. This complex is necessary for building the branched strands of the cell’s actin cytoskeleton, which allows them to change shape, divide, and move, amongst many other functions that allow cells to operate normally.
ARPC1B is most common in blood cells, which helps explain Daniel’s weak immune system and painful reactions to infections.
Hematologist Dr. Walter Kahr, lead author on the study, studied Daniel’s platelets, a type of blood cell. Kahr found that while normal platelets have a fried-egg shape, Daniel’s platelets are smaller and look more spider-like, with long spiky extensions. They lacked the ARPC1B protein completely.
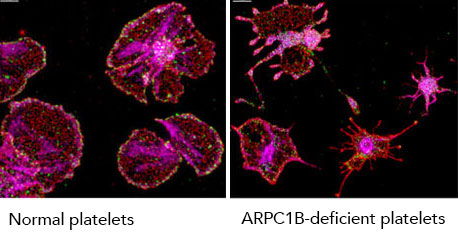
Normal platelets (left) have a fried-egg shape, and branched actin (purple). ARPC1B-deficient platelets, like Daniel’s, are smaller and have spider-like extensions.
Credit: Walter Kahr et. al., Nature Communications (cc-by-4.0)
Platelets are a key player in blood clotting, and Daniel’s spiky platelets help explain his bleeding problems.
Since discovering the mutation, the SickKids team also found two more patients within SickKids alone with a mutation on the same gene, and about 20 others worldwide.
A similar condition leading to misshapen platelets, Wiskott-Aldrich syndrome, is treated using bone marrow transplants. Doctors are hopeful that the same approach will work for Daniel, and others with ARPC1B mutations.
This new diagnosis ends the mystery, and marks the beginning of a treatment program that can address the root causes of illness for Daniel, and other patients around the world.